510(k) Einreichungsdienste
Der 510(k)-Weg ist ein Verfahren, das Hersteller von Medizinprodukten nutzen, um von der FDA die Freigabe für die Vermarktung ihrer Produkte in den USA zu erhalten. Dazu gehört die Einreichung einer Premarket-Benachrichtigung bei der FDA, die Details über das Design, die Leistungsmerkmale und den Verwendungszweck des Produkts enthält und wie diese mit einem zuvor zugelassenen „Prädikatsprodukt“ verglichen werden.
Das Ziel eines 510(k)-Antrags ist es, nachzuweisen, dass das neue Produkt „im Wesentlichen gleichwertig“ mit einem Prädikatsprodukt ist, das bereits von der FDA zugelassen wurde und kommerziell erhältlich ist. Die FDA prüft dann diesen Antrag, um diese Gleichwertigkeit zu bestätigen und sicherzustellen, dass das Gerät wie vorgesehen funktioniert.
Sobald die FDA bestätigt hat, dass das neue Gerät im Wesentlichen dem Prädikat entspricht, stellt sie ein Freigabeschreiben aus. Dies ermöglicht es dem Hersteller, das Gerät in den USA zu vermarkten. Es ist wichtig zu verstehen, dass eine 510(k)-Zulassung nicht dasselbe ist wie eine FDA-Zulassung, sondern vielmehr anzeigt, dass das Gerät einem bestehenden Gerät auf dem Markt ähnelt.
Die Standardgebühr für eine 510 (k) -Einreichung beträgt 21.760 USD, mit einer reduzierten Gebühr von 5.440 USD für kleine Unternehmen.
Definition der substantiellen Äquivalenz
Ein wichtiger Teil eines 510(k)-Antrags ist der Nachweis der wesentlichen Gleichwertigkeit mit einem legal vermarkteten Produkt in den USA. Das bedeutet, dass das neue Gerät genauso sicher und effektiv ist wie das Prädikatsgerät.
Ein Produkt gilt als im Wesentlichen gleichwertig, wenn es denselben Verwendungszweck wie das Prädikat und dieselben technologischen Merkmale aufweist oder wenn es denselben Verwendungszweck, aber unterschiedliche technologische Merkmale aufweist, die keine unterschiedlichen Fragen der Sicherheit und Wirksamkeit aufwerfen. Die bei der FDA eingereichten Informationen müssen zeigen, dass das Produkt genauso sicher und wirksam ist wie das legal vermarktete Produkt.
Es ist wichtig zu beachten, dass eine wesentliche Gleichwertigkeit nicht bedeutet, dass das neue Gerät mit dem Prädikatselement identisch sein muss. Die FDA prüft, ob der Verwendungszweck derselbe ist, und bewertet etwaige Unterschiede in den technologischen Eigenschaften, um sicherzustellen, dass sie keine neuen Sicherheits- oder Wirksamkeitsbedenken aufwerfen. Diese Bewertung umfasst eine Überprüfung der wissenschaftlichen Methoden, die zur Bewertung von Unterschieden und Leistungsdaten verwendet werden, wie z. B. klinische und nicht-klinische Labortests, Sterilität, Softwarevalidierung, Biokompatibilität usw.
Wenn die FDA feststellt, dass das Produkt nicht im Wesentlichen gleichwertig ist, hat der Antragsteller mehrere Möglichkeiten, darunter die erneute Einreichung eines 510(k)-Antrags mit neuen Daten, die Verfolgung einer De-Novo-Klassifizierung für die Klasse-I- oder II-Einstufung, die Einreichung eines Antrags auf Neueinstufung oder die Einreichung eines Zulassungsantrags vor dem Inverkehrbringen (PMA).
Arten von 510(k)s
Es gibt drei Arten von 510(k)s: Traditionell, Abgekürzt und Spezial. Es ist wichtig, die Qualifikationen für jeden Typ zu verstehen, um den geeigneten Weg zu wählen.
- Der traditionelle 510(k)- Weg wird für neue Produkte verwendet, die noch nicht freigegeben wurden und keinen PMA-Prozess (Pre-Market Approval) durchlaufen müssen. Es wird auch verwendet, wenn Änderungen an einem zuvor freigegebenen Gerät vorgenommen werden, das die Kriterien für ein Special 510(k) nicht erfüllt. Für diesen Weg ist es wichtig, Prädikatsprodukte hinsichtlich ihrer Anwendungshinweise und technologischen Merkmale zu identifizieren.
- Das Abbreviated 510(k)- Programm wurde als Alternative zum traditionellen Weg entwickelt. Hersteller können eine abgekürzte 510(k)-Datei einreichen, wenn sich ihre Einreichung auf FDA-Leitfäden stützt, die Einhaltung spezieller Kontrollen für den Produkttyp nachweist oder freiwilligen Konsensstandards entspricht. Eine abgekürzte 510(k)-Einreichung enthält die gleichen Abschnitte wie eine traditionelle 510(k)-Einreichung, aber Hersteller können ihre Einreichung mit zusammenfassenden Berichten zu Leitfäden, zur Einhaltung spezieller Kontrollen oder zur Konformität mit anerkannten Standards erweitern, um eine wesentliche Gleichwertigkeit nachzuweisen. Obwohl der abgekürzte Weg nicht unbedingt mit weniger Aufwand verbunden ist, kann er eine praktikable Option sein, wenn der Nachweis der Äquivalenz zu einem Standard einfacher ist als die Verwendung eines Prädikats. Der Überprüfungszeitraum für einen abgekürzten 510(k) beträgt in der Regel etwa 90 Tage, obwohl er in einigen Fällen länger dauern kann als ein traditioneller 510(k).
- Der Special 510(k) ist ein regulatorischer Weg, der für Änderungen an einem eigenen legal vermarkteten Prädikatsprodukt eines Herstellers konzipiert ist, das bereits für den kommerziellen Vertrieb zugelassen ist. Die jüngste Aktualisierung der Special 510(k)-Leitlinien ermöglicht die Überprüfung von Änderungen, die sich auf den Verwendungszweck eines Produkts auswirken oder seine grundlegende wissenschaftliche Technologie verändern, während solche Änderungen zuvor auf diesem Weg nicht zulässig waren.
Überprüfungsprozess und Zeitplan für die Einreichung von 510(k)
Das Verfahren für eine 510(k)-Einreichung umfasst mehrere Schritte:
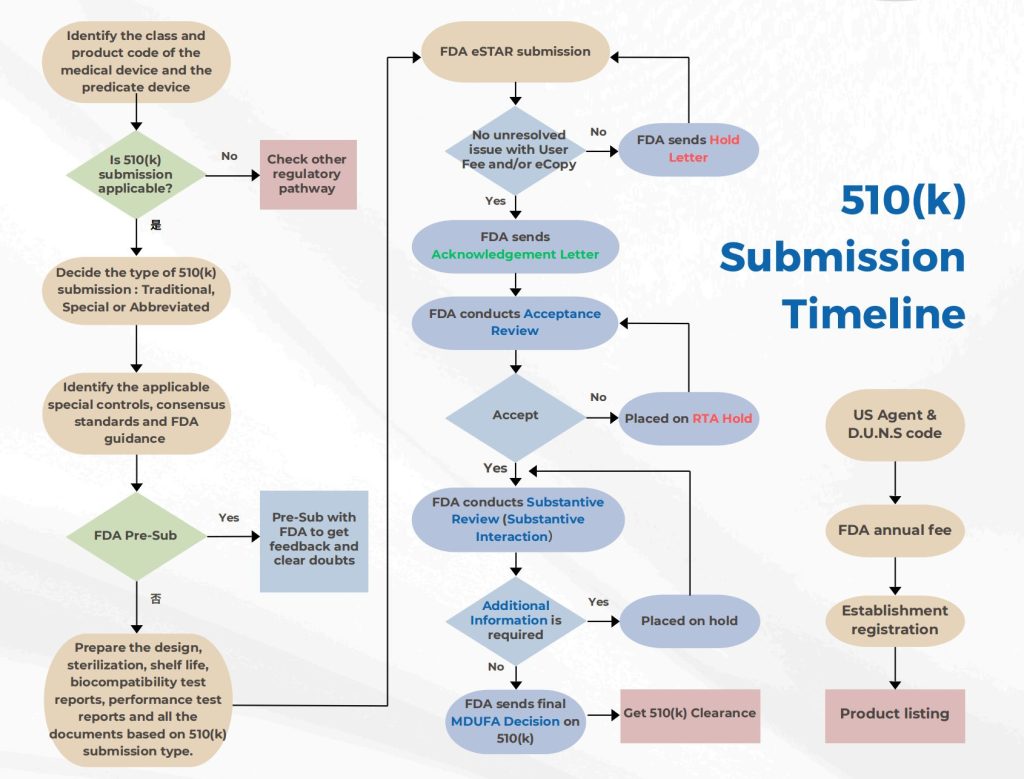
Hier ist der Zeitplan für die Überprüfung durch die FDA:
|
Tag 1 |
Innerhalb von 7 Tagen |
Bis Tag 15 |
|
Die FDA erhält den 510(k)-Antrag. |
Die FDA stellt in Fällen, in denen Bedenken hinsichtlich der Benutzergebühren oder der Einreichung einer eCopy bestehen, ein Bestätigungsschreiben oder ein Hold-Schreiben aus. |
Die FDA führt eine Annahmeprüfung durch, bei der sie den 510(k)-Antrag bewertet. Nach dieser Überprüfung informiert die FDA den Antragsteller, ob sein 510(k) zur inhaltlichen Prüfung angenommen oder in die RTA-Warteschleife (Refuse to Accept) gesetzt wurde. |
|
Bis Tag 60 |
Bis Tag 90 |
Bis Tag 100 |
|
Die FDA führt eine inhaltliche Prüfung durch. Sie teilen dem Antragsteller dann eine inhaltliche Interaktion mit, in der angegeben wird, ob die FDA mit einer interaktiven Überprüfung fortfahren oder zusätzliche Informationen anfordern wird. |
Die FDA sendet die endgültige MDUFA-Entscheidung zu 510(k). |
Wenn bis zum 100. Tag der Überprüfung keine MDUFA-Entscheidung (Medical Device User Fee Amendments) getroffen wird, gibt die FDA eine Mitteilung über die verpasste MDUFA-Entscheidung heraus. In dieser Mitteilung werden alle noch zu klärenden Überprüfungsfragen aufgezeigt. |
Wer muss einen 510 (k) einreichen?
Der FD&C Act und die 510(k)-Verordnung (21 CFR 807) legen nicht fest, wer einen 510(k)-Antrag einreichen muss. Stattdessen legen sie fest, für welche Maßnahmen, wie z. B. die Einführung eines Produkts auf dem US-Markt, eine 510(k)-Einreichung erforderlich ist.
Die folgenden vier Kategorien von Parteien müssen einen 510(k) bei der FDA einreichen:
Hersteller mit Sitz in den USA, die Geräte auf den US-Markt bringen, insbesondere wenn sie Geräte auf der Grundlage ihrer eigenen Spezifikationen entwickeln und vermarkten. Darüber hinaus wird Zubehör, das an Endverbraucher verkauft wird, als fertige Produkte behandelt und muss nach 510 (k) eingereicht werden. Hersteller von Gerätekomponenten sind jedoch davon ausgenommen, es sei denn, diese Komponenten werden als Ersatzteile an Endverbraucher verkauft. Vertragshersteller, die Produkte auf der Grundlage fremder Spezifikationen herstellen, sind ebenfalls ausgenommen. Spezifikationsentwickler, die Geräte auf dem US-Markt auf den Markt bringen. Diese Entwickler entwerfen Gerätespezifikationen, lassen sie aber im Rahmen von Verträgen herstellen. Sie sind für die 510(k)-Einreichung verantwortlich, nicht der Vertragshersteller. Umpacker oder Umetikettierer, die Geräteetiketten oder -bedingungen ändern, können eine 510(k)-Einreichung verlangen. Die meisten sind davon ausgenommen, aber wesentliche Änderungen wie das Hinzufügen neuer Verwendungen oder das Ändern von Warnhinweisen können eine Einreichung erforderlich machen. Ausländische Hersteller/Exporteure oder ihre US-Vertreter, die Produkte auf dem US-Markt einführen, müssen ebenfalls einen 510(k) bei der FDA einreichen. Darüber hinaus ist es wichtig zu beachten, dass alle Hersteller, einschließlich der Entwickler von Spezifikationen, von Geräten der Klassen II und III sowie von ausgewählten Geräten der Klasse I bei der Entwicklung ihrer Geräte die Designkontrollen einhalten müssen. Dazu gehört, dass die Dokumentation zur Designkontrolle für die FDA-Überprüfung während der Inspektionen zur Verfügung steht. Alle Änderungen an Produktspezifikationen oder Herstellungsprozessen müssen der Qualitätssystemverordnung (21 CFR 820) entsprechen und können eine neue 510(k)-Einreichung erforderlich machen.
Wann ist ein 510 (k) erforderlich?
Wenn Sie ein Produkt zum ersten Mal in den kommerziellen Vertrieb einführen, müssen Sie, sofern nicht ausgenommen, mindestens 90 Tage vor dem Anbieten des Produkts einen 510(k)-Antrag stellen. Diese Anforderung gilt für Produkte, die nach dem 28. Mai 1976 in Verkehr gebracht wurden, unabhängig davon, ob sie sich vor diesem Datum in der Entwicklung oder in der klinischen Prüfung befanden. Wenn Ihr Unternehmen das Gerät nicht vor dem 28. Mai 1976 vermarktet hat, ist eine 510(k)-Einreichung erforderlich.
Darüber hinaus muss der 510(k)-Inhaber feststellen, ob die Änderung oder Modifikation eines legal in Verkehr gebrachten Produkts erheblich beeinträchtigt werden könnte, wenn es sich um eine Änderung handelt, die wesentlich ist. Diese Entscheidung muss die Einhaltung der Qualitätssystemverordnung (21 CFR 820) berücksichtigen und im Gerätestammsatz und in den Änderungskontrolldatensätzen dokumentiert werden. Es ist ratsam, die Gründe für die Einreichung oder Nichteinreichung eines neuen 510(k) in den Änderungskontrolldatensätzen aufzuzeichnen.
Was sollte in einer 510(k)-Einreichung enthalten sein?
In der aktuellen elektronischen Einreichung von eSTAR 510(K) wurden die folgenden Informationen angefordert:
Art der Einreichung Anschreiben/Referenzschreiben Informationen zum Antragsteller Korrespondenz vor der Einreichung und frühere regulatorische Wechselwirkung Konsens Standards Produktbeschreibung Vorgeschlagene Indikation Klassifizierung Prädikate und wesentliche Gleichwertigkeit Design/Spezielle Kontrollen, Gesundheitsrisiken und Maßnahmen zur Risikominderung Kennzeichnung Aufbereitung Sterilität Haltbarkeit Biokompatibilität Software/ Firmware Cybersicherheit/ Interoperabilität Elektromagnetische Verträglichkeit (ECM), elektrisch, mechanisch, Leistungstests für drahtlose und thermische Sicherheit Referenzen Administrative Dokumentation Änderung/Zusätzliche Informationen (KI) Antwort.
Warum uns wählen?
Wenn es darum geht, die Komplexität der 510(k)-Einreichung zu bewältigen, zeichnet sich unser Team durch seine umfassende Erfahrung und sein Engagement für den Kundenerfolg aus. Wir haben eine nachgewiesene Erfolgsbilanz bei der Unterstützung von Unternehmen bei der Erlangung der FDA-Zulassung für ihre Medizinprodukte. Unser Ansatz integriert technisches, klinisches und regulatorisches Know-how, so dass wir Herausforderungen meistern und Ihre Markteinführungszeit verkürzen können. Wir verstehen die Feinheiten des 510(k)-Prozesses und können Sie durch jeden Schritt führen, von der Vorbereitung Ihrer Einreichung bis zur Beantwortung von FDA-Anfragen. Darüber hinaus bieten wir Übersetzungsdienste für wichtige Dokumente an, um sicherzustellen, dass Sie alle Informationen, die Sie benötigen, in einer Sprache haben, die Sie verstehen. Wählen Sie uns für Ihre 510(k)-Bedürfnisse und erleben Sie den Unterschied, den unser Wissen, unsere Erfahrung und unser kundenorientierter Ansatz ausmachen können.
Über Accel
Accel Groups ist ein engagiertes Team von erfahrenen Fachleuten in den Bereichen regulatorischer, klinischer und Marktzugang. Wir unterstützen Hersteller von Medizinprodukten und IVDs mit kompletten Lösungen für den Produktlebenszyklus, von der präklinischen, strategischen, klinischen Bewertung und Studie über die Einreichung von Zulassungsanträgen bis hin zur Überwachung nach der Markteinführung. Unsere regionalen Auftraggeber verfügen über mindestens 10+ Jahre Erfahrung in ihrem Fachgebiet, damit wir die regionale fundierte Expertise mit globalen One-Stop-Shop-Lösungen anbieten können. Fragen Sie uns nach unseren Start-up-Kits für Start-up-Unternehmen mit regulatorischen Pfaden in wichtigen Ländern.